La dispersión y diversificación de las poblaciones humanas modernas responde a múltiples factures, incluyendo selección natural, aislamiento reproductor, deriva genética, efecto fundador, etc. La aparición de los caracteres fenotípicos «raciales» responde a procesos de adaptación ambiental por selección natural en función de variables geográficas y climáticas que se distribuyen geográficamente formando clinas (variaciones graduales de la frecuencia o expresión de un carácter), por lo que la frecuencia y la expresión fenotípica de dichos caracteres distribuye formado un gradiente continuo de variabilidad, sin que existan grupos discretos y aislados desde el punto de vista biológico. Las barreras culturales determinan una diversificación poblacional que generalmente es superior a las diferencias biológicas. Aun así, las sociedades humanas tradicionales presentan una gran variabilidad morfológica que es posible caracterizar y que deriva de su historia evolutiva y dinámica poblacional particulares. Los procesos evolutivos y de diversificación genética que han actuado en cada momento y en cada zona geográfica, y su velocidad, han variado enormemente. El análisis de la diversidad racial humana actual permite caracterizar qué procesos han actuado en cada momento, con el fin de entender el origen de nuestra diversidad biológica.
La diversidad morfológica responde generalmente a factores adaptativos, por lo que la expresión fenotípica y la frecuencia de dichos caracteres depende de la presión selectiva del medio. A largo plazo, las adaptaciones humanas dependen de las condiciones de cada ambiente. Las principales adaptaciones humanas (a la temperatura, radiación solar, altitud, humedad relativa) que se describieron en temas anteriores son la fuente de la mayor parte de la diversidad morfológica de las poblaciones humanas actuales. En este capítulo analizaremos la diversidad molecular humana, que no tiene reflejo en la variabilidad morfológica, y los factores que la determinan.
Grupos sanguíneos
El sistema ABO
El sistema sanguíneo ABO es uno de los numerosos polimorfismos de la sangre humana. Los antígenos de los hematíes (glóbulos rojos) han sido marcadores genéticos útiles en estudios familiares, poblacionales y en análisis de ligamiento a causa de su fácil caracterización, su herencia mendeliana y la gran variabilidad de las frecuencias alélicas que se puede dar en las diferentes poblaciones. El ABO y el RH son los polimorfismos proteicos más importantes en la transfusión sanguínea y el trasplante de órganos y tejidos. El descubrimiento de los grupos sanguíneos ABO proviene de la observación inicial de Landsteiner (1901) de una posible aglutinación entre diversas muestras de sangre humana, concretamente entre el suero y los glóbulos rojos. De ello dedujo la existencia de cuatro tipos sanguíneos de acuerdo con la presencia de dos antígenos, A y B, sobre la superficie de los glóbulos rojos y la presencia de los correspondientes anticuerpos, anti-A y anti-B, en el plasma. Existen cuatro fenotipos posibles: O, A, B, y AB. Las personas del tipo A tienen el antígeno A en sus glóbulos rojos, las del tipo B tienen el antígeno B, las del AB tienen ambos antígenos A y B, y las del tipo O no tienen ninguno. Así, la reacción de los glóbulos rojos de cada tipo con anticuerpos anti-A y anti-B es la siguiente:


Determinantes antigénicos del sistema ABO
El suero no contiene nunca el anticuerpo que reaccionaría con los hematíes de la propia sangre (esto es una regla válida para todos los sistemas de grupos sanguíneos). La presencia constante de los anticuerpos anti-A y anti-B confiere al sistema ABO una característica particular y ha sido la causa de que este sistema de grupos sanguíneos haya sido descubierto en primer lugar. Los anticuerpos del sistema ABO han sido llamados anticuerpos regulares y constituyen el principal riesgo de la transfusión sanguínea. En general son potentes y reaccionan no sólo a temperatura ambiente, sino también a 37° C, la temperatura del cuerpo humano. Del cuadro anterior se deduce que el grupo O será el donante universal y el grupo AB el receptor universal. En algunos casos los anticuerpos del grupo O (anti-A y anti-B) son muy potentes y pueden dar problemas transfusionales, estos son los llamados O peligrosos. Los anticuerpos anti-A y anti-B, regularmente presentes en el adulto, no lo están necesariamente en el recién nacido ni durante los primeros meses de vida. El recién nacido no produce todavía sus propios anticuerpos. Sí existen anticuerpos en su suero proceden de la madre son generalmente de escasa potencia ya que, atraviesan la placenta con dificultad. Así, un recién nacido del grupo O, de madre A (no poseerá anticuerpos anti-A), podrá tener anti-B, pero en general será de reacción antigénica débil.
Antígenos ABH
 El sistema ABH es en realidad el mismo sistema ABO pero analizado a un nivel superior considerando otros loci que afectan a la expresión de los alelos A y B. La presencia de los antígenos A, B ó O en los hematíes depende de la herencia de los alelos A, B y O. Además, existe un gen H (del ABH), situado en un locus separado, que codifica el enzima α-2-L-fucosiltransferasa. modificador de la sustancia precursora H (sustancia H del ABO) sobre la que actúan los productos (enzimas) de los genes A y B (transferasas). La sustancia H es modificada por las transferasas codificadas por los genes A y B, que son enzimas que convierten la substancia H en antígeno A o B, respectivamente. El fenotipo O es silencioso, no altera la estructura de la sustancia precursora H (por ausencia de transferasas); por tanto, los individuos del grupo O tienen grandes cantidades de sustancia H en sus células; los A y B también tienen pero menos cantidad. Este gen H del ABH (con dos alelos H y h) se detectó al comprobar que en determinados casos no se expresaban los grupos sanguíneos A y B. Ello se debe al genotipo hh (homozigoto del ABH), por lo que al no existir producto del alelo H no es posible la transformación del precursor en sustancia H. Además el genotipo hh modifica la expresión fenotípica de los antígenos A, B y H y altera también el Secretor (Se) y el Lewis (Le).
El sistema ABH es en realidad el mismo sistema ABO pero analizado a un nivel superior considerando otros loci que afectan a la expresión de los alelos A y B. La presencia de los antígenos A, B ó O en los hematíes depende de la herencia de los alelos A, B y O. Además, existe un gen H (del ABH), situado en un locus separado, que codifica el enzima α-2-L-fucosiltransferasa. modificador de la sustancia precursora H (sustancia H del ABO) sobre la que actúan los productos (enzimas) de los genes A y B (transferasas). La sustancia H es modificada por las transferasas codificadas por los genes A y B, que son enzimas que convierten la substancia H en antígeno A o B, respectivamente. El fenotipo O es silencioso, no altera la estructura de la sustancia precursora H (por ausencia de transferasas); por tanto, los individuos del grupo O tienen grandes cantidades de sustancia H en sus células; los A y B también tienen pero menos cantidad. Este gen H del ABH (con dos alelos H y h) se detectó al comprobar que en determinados casos no se expresaban los grupos sanguíneos A y B. Ello se debe al genotipo hh (homozigoto del ABH), por lo que al no existir producto del alelo H no es posible la transformación del precursor en sustancia H. Además el genotipo hh modifica la expresión fenotípica de los antígenos A, B y H y altera también el Secretor (Se) y el Lewis (Le).
El fenotipo Bombay (Oh) se da en los individuos que no heredan un gen H (genotipo hh)
Bombay (Oh). Estos individuos no producen substancia H y, por tanto, los genes A y B no pueden expresarse. El fenotipo Bombay (Oh) es muy raro (1/106) pero en la población Marathi de Bombay es de 1/13.000, con una frecuencia alélica de f(h) = 0.0066. Los individuos Bombay pueden heredar los genes A o B, pero debido a su carencia de substancia H no pueden producir ni el antígeno A ni el antígeno B. Los hematíes del fenotipo Bombay son aparentemente de fenotipo O, pero a diferencia de éstos, que tienen gran cantidad de antígeno H, los hematíes Bombay carecen de dicha substancia. Los hijos de individuos Bombay pueden expresar cantidades normales de los antígenos A y/o B en sus hematíes siempre que hereden un gen H del otro progenitor; de este modo, dos individuos cuyo fenotipo es O en apariencia pueden tener un descendiente con un fenotipo A.
Subgrupos de A y de B
El fenotipo A puede dividirse en dos subgrupos. Aproximadamente el 80% de los individuos del grupo A tienen el fenotipo A1 y el 20% el fenotipo A2. Entre estos dos subgrupos existen diferencias cualitativas y cuantitativas.
Existen diversos tipos de cadenas de sustancia precursora H. Los individuos A1 producen antígeno A a partir de todas las cadenas H de tipo II (H1, H2, H3, H4). Los individuos A2 producen antígeno A solamente a partir de precursores H1 y H2. Por tanto, los individuos A1 presentan más cantidad de antígeno A por hematíe que los individuos A2. Aproximadamente el 3% de los individuos A2 y el 25% de los individuos A2B producen un anticuerpo denominado anti-A1. Este anticuerpo reacciona con los hematíes A1, pero no reacciona con los A2. Otros hematíes pertenecientes al grupo A3 presentan un modelo característico de aglutinación cuando reaccionan con el suero anti-A: algunos de los hematíes son aglutinados mientras que otros no (dan una imagen de doble población). El fenotipo A3 presenta una frecuencia de 1‰. Otros subgrupos raros de A son, Ax, Am, Aend, Ael y AFinn. Es importante la identificación de los donantes de sangre que pertenecen a estos subgrupos débiles de A con objeto de evitar que sean erróneamente etiquetados como donantes del grupo sanguíneo O. Si se transfundiera sangre de uno
de los subgrupos de A indicados a un receptor de grupo O podría tener lugar una reacción transfusional. Los subgrupos de B son menos comunes. Entre ellos tenemos B3, Bx, Bm o Bel.
Anticuerpos del sistema ABO
Los anticuerpos anti-A y Anti-B son producidos por individuos que carecen de los respectivos antígenos A y B. Estos anticuerpos son en su mayoría inmunoglobulinas de tipo IgM. También pueden ser IgG, pero son menos frecuentes, y generalmente son producidos por individuos del grupo O. El anticuerpo anti-AB lo producen individuos pertenecientes al fenotipo O. El anticuerpo anti-AB no es una mezcla de anti-A y anti-B, sino un tercer anticuerpo que presenta reacción cruzada con un antígeno presente en ambos hematíes A y B. Este antígeno se denomina compuesto AB o antígeno C. Los anticuerpos del sistema ABO pueden reaccionar a la temperatura corporal (37°C) y activar el complemento causando una rápida destrucción intravascular de los hematíes. El título (nivel o sensibilidad) de la aglutinación de anti-A y de anti-B con frecuencia está disminuido en las personas ancianas y en los pacientes con hipogammaglobulinemia. Los niños recién nacidos no producen anti-A ni anti-B hasta, que se empiezan a formar hacia los 3-6 meses de edad. El anti-A1 es un anticuerpo natural de tipo IgM que producen algunos individuos A2 y A2B. El anti-A1 generalmente tiene un rango térmico bajo, por lo que no suele tener significación clínica. Los individuos A2, cuyo suero contiene anti-A1, con un rango térmico lo bastante elevado para tener significación clínica deben ser transfundidos con sangre del grupo O o A2. El anticuerpo anti-H puede presentarse como un auto-anticuerpo natural en el suero de individuos A1 o A1B o en el plasma de individuos del fenotipo Bombay. En este caso, su rango térmico es elevado, lo cual, junto con su capacidad para fijar el complemento, hace que el alo-anticuerpo anti-H sea clínicamente significativo. Así pues, los individuos Bombay solamente pueden ser transfundidos con sangre de otros individuos pertenecientes a dicho fenotipo.
Pseudoalelismo de A y B o AB cis
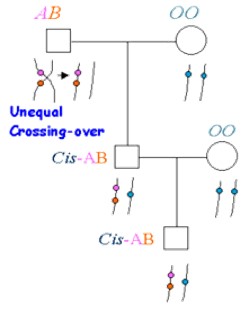
Pseudoalelismo AB cis
Si A y B son alelos verdaderos, no pueden ser transmitidos simultáneamente. La regla es que las parejas AB × O no pueden tener hijos AB. Esto es cierto en la mayoría de los casos pero se ha observado un número muy reducido de familias en las que los genes A y B son se encuentran en el mismo cromosoma. Así, se puede dar que O × AB = AB. La hipótesis más plausible para explicar esto sería que en realidad A y B no serían alelos de un solo locus sino loci muy próximos en el mismo cromosoma con ausencia casi completa de entrecruzamiento. Entonces A y B serían pseudoalelos: A (que transmite A) y a (que no transmite A), B (que transmite B) y b (que no trasmite B). En la mayoría de casos las combinaciones que se producen son Ab, aB y ab. Estas combinaciones no se distinguen de la serie tradicional A, B y O. Las reglas permanecen pues válidas a causa de la ausencia de entrecruzamiento. La combinación AB no se observa casi nunca. El examen de los individuos que pertenecen al grupo AB, permite sospechar si pertenecen a la combinación denominada AB cis (AB/ab) o AB normal (Ab/aB).
Distribución racial del ABO
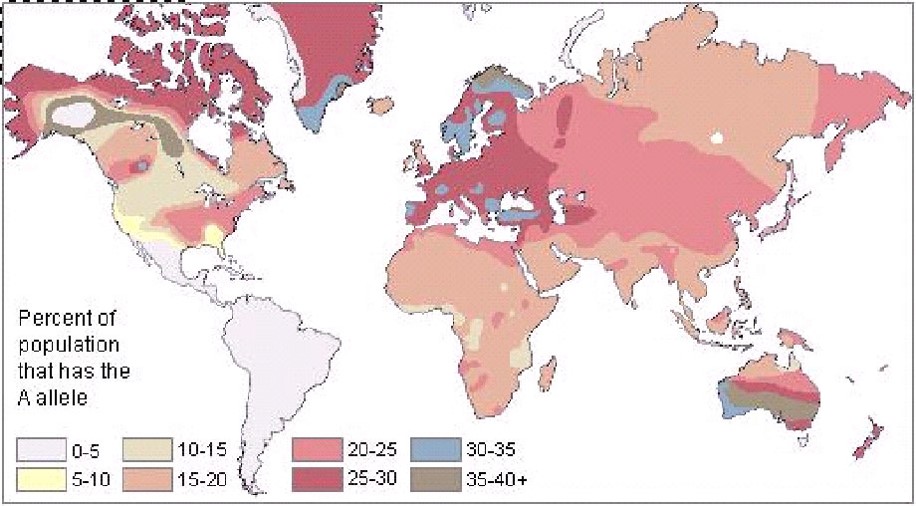
La frecuencia del alelo O oscila entre 0,4 y 1; la del A entre 0 y 0,55, y la del B entre 0 y 0,3. El grupo O es prácticamente el único en poblaciones amerindias no mestizadas de Sudamérica, Centroamérica y parte meridional de Norteamérica; solo entre los esquimales baja su frecuencia. Núcleos de frecuencia alta del grupo O se encuentran en los sardos, los vascos y beréberes del Atlas, quizá debido a su aislamiento
genético o por efecto fundador.

La frecuencia del alelo A oscila en Europa entre 0,25 y 0,30, con frecuencias máximas en lapones, que presentan los valores más altos del subgrupo A2, y armenios. También es alta entre los esquimales y en algunos grupos de nor-amerindios canadienses, donde alcanza su valor más elevado; y el grupo A es también muy abundante en Australia y Asia oriental, así como en las poblaciones polinesias.
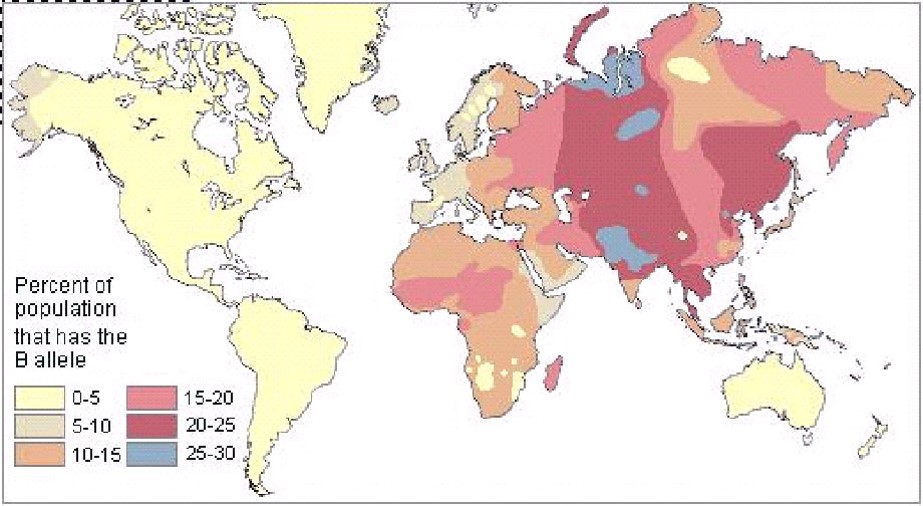
El grupo B es frecuente en poblaciones del Himalaya (q = 0,30) y en cambio desaparece en los amerindios y en los Aborígenes Australianos. Desde Asia central y oriental su frecuencia va disminuyendo tanto hacia Melanesia y Polinesia como hacia Africa, y en sentido occidental en dirección a Europa, donde alcanza los valores mínimos entre los vascos.
Los antígenos ABH en secreciones: el gen secretor
Los antígenos eritrocitarios A, B y H se hallan también en el plasma, en la saliva y en otros líquidos orgánicos. Su presencia en esos líquidos está controlada por un alelo del gen secretor (Se) que es autosómico dominante. Las substancias A, B y H se hallan en el plasma de todos los individuos independientemente de que sean secretores o no, pero son mucho más abundantes en el plasma de los secretores. El 80% de los individuos de raza blanca heredan el gen Se (secretores SeSe o Sese). El 20% restante no son secretores (genotipo sese). No se ha identificado ningún producto específico del alelo Se, aunque se sabe que regula la formación de substancias H en las secreciones y en el plasma. Así, un secretor del grupo A tiene substancias A y H en sus líquidos orgánicos y los no secretores (sese) no. La determinación de secretores (se suele hacer en saliva) tiene escasa importancia tanto clínica como de laboratorio, pero puede ser útil en la determinación del grupo sanguíneo ABO de un individuo si el grupo determinado en sus hematíes no es concluyente.
El sistema Lewis (Le)
Los antígenos del sistema Lewis proceden del plasma y se adsorben a los hematíes. Los alelos del sistema Lewis se representan por los símbolos Le y le; Le representa el alelo dominante y le el recesivo. El gen Le codifica un enzima responsable de la adición de fucosa (Fuc) subterminal a una cadena de tipo I de substancia precursora H. La fucosa no puede añadirse a las cadenas de tipo II de los hematíes. Por ello, los antígenos de sistema Lewis no forman parte de la membrana de los hematíes, sino que son substancias adsorbidas procedentes del plasma. Se han descrito tres fenotipos Lewis comunes: Le(a+b−), Le(a−b+) y Le(a−b−), que son el resultado de la interacción de los genes Le, H y Se.

Adición subterminal de una Fucosa por el enzima codificado por el alelo Le del sistema Lewis.
Los antígenos Lea y Leb
La estructura bioquímica de los antígenos Lea y Leb difieren solamente en una molécula de fucosa. El gen H añade fucosa a la galactosa terminal de la substancia precursora del plasma si el individuo es secretor. La substancia resultante es el antígeno H. Los no secretores no añaden la molécula de fucosa, aunque hereden el gen H, y de este modo no se altera la sustancia precursora en el plasma. Esta fucosa se añade en todos los individuos que heredan el alelo Le independientemente de si son o no secretores. Pero los no secretores que heredan los genes H y Le añaden una sola molécula de fucosa subterminal. Esta estructura es reconocida como antígeno Lea. Los secretores que heredan el gen H y el gen Le añaden dos moléculas de fucosa a la substancia precursora. El producto resultante es conocido como antígeno Leb. Los hematíes de los individuos no secretores que heredan el alelo Le presentan el fenotipo Le(a+b−), mientras que los secretores que poseen el gen Le presentan el fenotipo Le(a−b+). Los individuos que carecen del alelo Le (6% de los individuos de raza blanca y 22% de los negros) pertenecen al fenotipo Le(a−b−). Dependiendo de si son secretores o no, pueden tener substancia H en su plasma.
Anticuerpos anti-Lewis
Los anticuerpos anti-Lea y anti-Leb son alo-anticuerpos naturales de clase IgM producidos por algunos individuos Le(a−b−). Ocasionalmente el anti-Leb es producido por individuos cuyo fenotipo es Le(a+b−). Los individuos Le(a−b+) no producen anti-Lea, dado que poseen en su plasma pequeñas cantidades de antígeno Lea. Los anticuerpos del sistema Lewis pueden aglutinar los hematíes y activar el complemento in vitro, pero in vivo tienen escasa importancia por dos razones: 1) los antígenos solubles Lea y Leb presentes en el plasma del donante neutralizan los anticuerpos del receptor, y 2) los antígenos Lewis son rápidamente desplazados de los hematíes del donante, que se transforman y adquieren un fenotipo Lewis igual al del receptor. Los anticuerpos anti-Lewis que solamente reaccionan a temperatura ambiente no tienen significación clínica. Los pacientes que presentan algún anticuerpo anti-Lewis que reacciona a temperatura superior a los 30°C o produce hemolisis in vitro, deben recibir sangres compatibles, asegurándose de ello mediante prueba cruzada.

Interacción entre los alelos H (precursor), Le (Lewis) y Se (Secretor).
El sistema Rhesus (RH)
El sistema Rhesus está constituido por unos 40 antígenos distintos, cinco de los cuales revisten una importancia especial. Para designar los diferentes antígenos del sistema Rh existen dos nomenclaturas principales que se utilizan indistintamente:
- Fisher-Race: distingue 3 genes situados en loci muy próximos. Los 5 alelos más comunes que pueden ocupar dichos loci se designan con los símbolos D, d, C, c, e. Cada alelo (con excepción del d) codifica un antígeno específico, que puede ser detectado en la membrana del hematíe. La presencia o ausencia del antígeno D es la que determina si un individuo es Rh positivo o negativo.
- Wiener: supone un solo gen de cada progenitor con una estructura en mosaico, que comprendería un número variable de antígenos sanguíneos. Así el gen R1 codificaría factores que corresponderían a los alelos C, D y e de Fisher-Race. El gen r produciría los antígenos c y e pero no D, C ni E.

El sistema sanguíneo del RH. El alelo D determina el RH+ y su ausencia el RH-.

Genotipos del sistema RH.
El 85 % de los individuos de raza blanca expresan el antígeno D y se les denomina Rh positivos. Si el antígeno D no se expresa, se denomina al individuo Rh negativo. El genotipo de la mayoría de individuos Rh negativos es rr (dce/dce). Los individuos que heredan los antígenos C o E, pero no el D, se clasifican como Rh negativos (por ejemplo dCe/dce). El antígeno D es un potente inmunogénicos. Aproximadamente el 70 % de los individuos Rh – producen anti-D si reciben sangre Rh +. Debido a que los antígenos C y E no son tan inmunogénicos como el D, no se determinan en las pruebas de rutina. La terminología de Fisher y Race se emplea para describir los antígenos Rh, mientras que la notación de Wiener es usada para describir un genotipo Rh.
Variante Du
Du es una variante débil del antígeno D, poco frecuente entre los individuos de raza blanca, pero común entre los individuos de raza negra (22%). Los hematíes Du generalmente dan reacciones débiles o negativas con el anticuerpo anti-D, siendo generalmente detectados gracias a la prueba indirecta de la antiglobulina (prueba Du). Los hematíes Du pueden ser clasificados en tres categorías:
- Du deprimido: la herencia del alelo C en posición trans con relación al D (dCe/Dce) tiene como resultado una expresión débil del antígeno D en los hematíes (Du). Los individuos que presentan estas características no producen anti-D si reciben sangre Rh positiva.
- Variante Du. El antígeno D presenta una estructura que consta como mínimo de cuatro partes. Si falta una o más partes del antígeno, el resto de éste puede tener una expresión débil. Un individuo con la variante Du puede producir alo-anticuerpos contra la parte de antígeno D que le falta. Por esta razón, los individuos que pertenecen a dicha variante deben ser transfundidos con sangre Rh negativa. Los centros de transfusión sanguínea efectúan la prueba para el factor Du a todos sus donantes Rh negativos, ya que la sangre de un Du inyectada a un receptor Rh negativo puede producir en este último una sensibilización al antígeno D.
- Du hereditario. Algunos individuos Du no pueden ser clasificados como Du deprimido ni como variante Du, puesto que aunque poseen el antígeno D completo, éste se expresa débilmente, desconociéndose la causa.
Hematíes con delección Rh (−D−) y Rh nulo
Los hematíes que no poseen antígenos dependientes de los loci C ni E se dice que presentan delección en el sistema (−D−). El número de lugares antigénicos D está muy aumentado en estos hematíes, y por tanto en anticuerpo anti-D de tipo IgG puede aglutinarlos. Los individuos que no expresan ninguno de los antígenos del sistema Rhesus en sus hematíes tienen Rh nulo (− − −): los hematíes de estos individuos tiene alterado el transporte de sodio y potasio. Esto da lugar a una anemia hemolítica caracterizada por estomatocitosis, esferocitosis y aumento de la fragilidad osmótica.
Anticuerpos del sistema Rhesus
Los anticuerpos del sistema Rhesus son casi siempre IgG (algunos sueros contienen cierta proporción de IgM e incluso de IgA), siendo estimulada su producción por transfusión o por embarazo. No activan el complemento debido a que la situación de los antígenos Rhesus en la membrana del hematíe no permite la formación de dobletes de IgG necesarios para su activación. Los anticuerpos del sistema Rh pueden causar reacciones transfusionales y enfermedad hemolítica del recién nacido.
Proteínas plasmáticas
Haptoglobinas

Polimorfismo de las haptoglobinas.
Estas proteínas plasmáticas son glicoproteínas que constituyen aproximadamente el 20 % de la fracción α2-globulina y se hallan en una cantidad promedio por individuo de 100 mg / 100 ml de suero. Su función es combinarse con la hemoglobina producida por la lisis de los eritrocitos, impidiendo que se pierda por excreción renal y conservando de esta forma el hierro. Las primeras observaciones sobre el comportamiento electroforético de las haptoglobinas llevaron a la conclusión de que existían tres fenotipos comunes controlados por dos alelos codominantes en un locus autosómico con dos variantes alélicas principales (Hp1, Hp2), por lo que los genotipos posibles son homozigoto Hp 1-1 (Hp1Hp1), heterozigoto Hp 2-1 (Hp1Hp2) y homozigoto Hp 2-2 (Hp2Hp2). Estos tres fenotipos se encuentran en las poblaciones humanas con frecuencias muy distintas. Junto con estos fenotipos electroforéticos comunes se pueden encontrar individuos con variantes raras que se deben a la existencia de alelos de muy baja frecuencia. La molécula de haptoglobina es un tetrámero que consiste en dos cadenas polipeptídicas α y dos β. Salvo excepciones, las cadenas β son idénticas en todas las personas.
Las cadenas α pueden ser de tres tipos que varían en sus propiedades de movilidad electroforética. El fenotipo Hp 1-1 puede dar lugar a cadenas α muy rápidas (cadenas llamadas hp1F), a cadenas lentas (hp1S) o a una mezcla de ambas. El fenotipo Hp 2-2 origina una sola clase de cadena α, mucho más lenta que las dos anteriores (hp2). El fenotipo Hp 2-1 heterozigoto, produce bien las bandas Hp2 combinadas con Hp1F, o bien esa misma banda combinada con Hp1S. El fenotipo Hp 2-1 modificado presenta una reducción en la intensidad de la banda Hp2 (Hp2mod). Estudios genealógicos mostraron que esta variabilidad depende de los correspondientes alelos (Hp1F, Hp1S, Hp2, Hp2mod). El genotipo Hp2modHp2mod es el fenotipo Hp0. El polimorfismo de las haptoglobinas parece ser evolutivamente muy antiguo, porque los alelos Hp1 y Hp2 se han encontrado en todas las poblaciones humanas estudiadas. El análisis de las frecuencias alélicas encontradas en poblaciones representativas de los principales grupos humanos revela una amplitud variación de Hp1, que oscila entre 0,7-0,.8 en algunos negroides, melanesios y sudamerindios, y menos de 0,1 en algunos hindúes (tamiles e irulas). En Europa el promedio de Hp1 es de 0,37-0,41, con un mínimo en el Levante peninsular y máximo entre los vascos. Existe una clina para este alelo en Europa con frecuencias altas en el Sur. En Asia el alelo Hp1 se da con frecuencias bajas (<0,25). Por contra, los Melanesios de Nueva Guinea presentan valores superiores a 0,7. También hay una clina NS en el América, de 0,3 al N hasta 0,7 al Sur. En África, los Pigmeos, Bosquimanos y Hotentotes tienen frecuencias altas (quizá sean las más antiguas).
Transferrina

Captación y transporte de Fe por las Transferrinas.
La presencia en el plasma humano de una proteína portadora de hierro fue descubierta por Holmberg y Laurell (1945) y por Schade y Carolina (1946). La transferrina (Tf) es una glucoproteina de la fracción β-globulina, tanto del suero humano como el de otros vertebrados. En 1957, Smithies describió la existencia de variantes genéticas de transferrina utilizando electroforesis en gel de almidón. Actualmente se conocen numerosas variantes Tf de interés antropológico. La función principal de esta proteína es mantener en solución y transportar el hierro férrico procedente de la absorción intestinal y de sus lugares de depósito (hígado, bazo, etc.), hasta la médula ósea y otros lugares del retículo endotelial, donde se utiliza para la síntesis de proteínas que contienen hierro. Además participa en la regulación y control de la absorción de hierro e impide la pérdida en el riñón. También actúa como tampón que previene la intoxicación por hierro y minimiza los cambios de actividad férrica en el plasma, ya que en condiciones normales solo una tercera parte de la transferrina sérica se halla saturada por dicho metal. Asimismo se ha comprobado que la transferrina puede tener un efecto bacteriostático ya que debido a su capacidad de unirse al hierro libre de la sangre, puede competir por este metal con la mayoría de los microorganismos, constituyendo así parte de lo que se denomina “inmunidad nutricional” (Weinberg, 1974). A este respecto, todos los estudios de inhibición del crecimiento de Klebsiella pneumoniae para distintas variantes de transferrina humana demuestran que existe una gran variabilidad en su actividad bacteriostática. La variante TfC es más efectiva que la TfD. La variante normal (1 banda en gel) es la TfC, que se puede desdoblar en dos: TfB (+móbil) y TfD (- móbil). Se dan combinaciones de todo tipo. La distribución de variantes de Tf es variable. En európidos 1% TfBC, 99% TfC; mongoles y negroides 5% TfCD, 95% TfC y raros TfD.
Inmunoglobulinas
Aparecen como respuesta a un alo-antígeno. Son anticuerpos del sistema inmunitario, tanto solubles como ligados a la membrana de los linfocitos B. Las Ig se componen de 4 cadenas polipeptídicas: 2 pesadas H y 2 ligeras L. Ambas constan de una región constante C y otra variable V.

α-1 Antitripsina
La Deficiencia de α-1 Antitripsina (α-1) es un trastorno genético hereditario que puede ocasionar en la tercera y cuarta década de vida una enfermedad pulmonar obstructiva crítica (EPOC) como enfisema y bronquitis. Con menos frecuencia se puede manifestar desde el nacimiento hasta cualquier momento en la vida como una enfermedad hepática. Los individuos afectados se caracteriza por unos niveles en la sangre muy bajos o inexistentes de una proteína llamada AAT (α-1 antitripsina) que es producida por el hígado. La función principal de la AAT es proteger el tejido pulmonar de la inflamación ocasionada por las infecciones y los irritantes inhalados, como el humo del tabaco. Los bajos niveles de AAT en sangre ocurren porque el hígado no puede liberar la AAT defectuosa a la rapidez normal. En una proporción pequeña de los afectados, la acumulación de la AAT defectuosa ocasiona daño grave al hígado.
Hay cinco genotipos posibles formados a partir de seis alelos (M, M1, M2, M3, Z, S). M es el alelo normal y los alelos M1, M2 y M3 afectan a exones del gen, por lo que no afectan a la estructura de la proteína. En cambio, las mutaciones Z y S se localizan en intrones del gen y alteran la estructura de la proteína resultante (variantes estructurales), por lo que pueden causar afectaciones patológicas.
- Normal (MM): No tiene el trastorno i no es portador de genes AAT deficientes.
- Portador (MZ): Deficiencia de AAT leve o moderada. Podría desarrollar la enfermedad y es portador de un alelo AAT alterado.
- Portador (MS): No está claro si existe riesgo de desarrollar síntomas de la enfermedad. Es portador de un alelo AAT alterado.
- α-1 (ZZ ó SZ): Deficiencia de AAT moderada a severa, podría desarrollar síntomas de enfermedad y es portador de dos alelos AAT alterados.
- α-1 (SS): No está claro si tiene riesgo de desarrollar síntomas de la enfermedad. Es portador de dos gens AAT alterados.
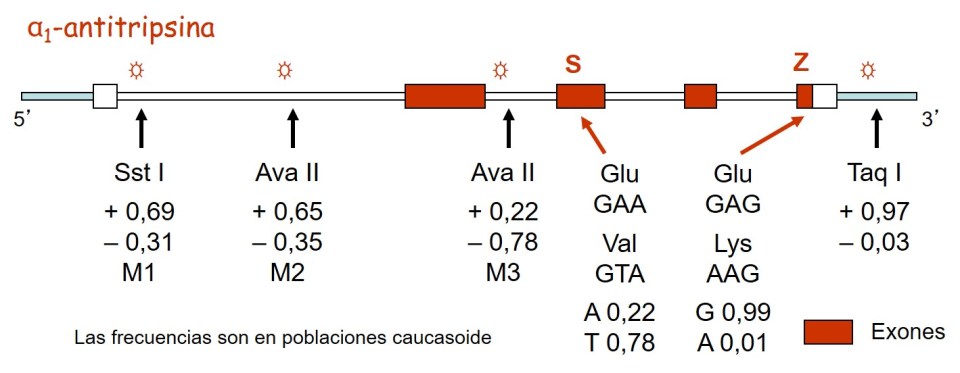
 Las frecuencias de los alelos varía entre poblaciones humanas. La variante M1 es más frecuente en poblaciones africanas que en otras regiones y en portugueses su frecuencia es baja. Las variantes S y Z son poco frecuentes por la acción de la selección natural, pero aún así se han mantenido en las poblaciones probablemente por presentar alguna asociación con otros genes que pueden conferir resistencia a enfermedades infecciosas.
Las frecuencias de los alelos varía entre poblaciones humanas. La variante M1 es más frecuente en poblaciones africanas que en otras regiones y en portugueses su frecuencia es baja. Las variantes S y Z son poco frecuentes por la acción de la selección natural, pero aún así se han mantenido en las poblaciones probablemente por presentar alguna asociación con otros genes que pueden conferir resistencia a enfermedades infecciosas.
Polimorfismos del HLA del MHC
El sistema polimórfico de antígenos leucocitarios humanos (HLA, Human
Leucocyte Antigen) forma parte del Complejo Mayor de Histocompatibilidad (MHC, Major Histocompatibility Complex) humano (en el ratón se denomina sistema H-2). Su función se enmarca en la respuesta ante infecciones por las moléculas receptoras del sistema inmunológico humano:
- Las inmunoglobulinas, secretadas por las células B
- Los receptores de las células linfocitarias T, TCR (T Cell Receptors formados por 2 cadenas: α i β ó γ i δ (codificadas en el cromosoma 14 las α i β; y en el cromosoma 7 las γ y δ) de las células linfocitaria citotóxicas TC o las cooperadoras (helpers) TH.
- Las glicoproteínas del sistema HLA del MHC, moléculas de Clase I y Clase II, dedicadas a la supresión de la infección y al mantenimiento de la integridad de los tejidos celulares gracias a su especificidad y gran variabilidad.
El sistema genético del HLA humano es el más variable de los polimorfismos estructurales conocidos y tiene gran importancia clínica en el trasplante de tejidos y en la respuesta inmune ante procesos cancerosos, infecciosos o depresores
del sistema inmunitario. Cuando un patógeno invade el organismo, las células inmunitarias (linfocitos B) segregan anticuerpos (inmunoglobulinas) que se unen a las aloproteinas o alovirus desactivándolos. En cambio las células linfocitaria T intervienen en la presentación antigénica por parte de las glicoproteínas del sistema HLA, bien posibilitando la lisis de las células infectadas por las células TC (citotóxicas) o estimulando la secreción de anticuerpos por las células TH (helpers). La conformación somática de los genes de las inmunoglobulinas y los receptores TCR limitan a las células linfocitarias B y T a una única especificidad antigénica pero el conjunto de células del sistema inmune posee millones de especificidades. Los receptores de las inmunoglobulinas segregadas por los linfocitos B reconocen los antígenos proteicos nativos (aloantígenos sin modificar) mientras que los receptores TCR de los linfocitos T sólo reconocen fragmentos peptídicos pequeños unidos a las glicoproteinas polimórficas del sistema HLA.
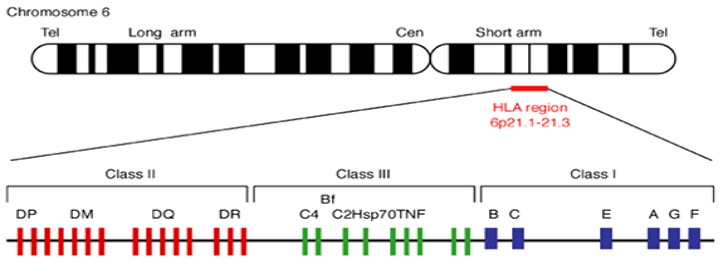
Estructura genética
El sistema MHC humano se localiza en el brazo corto de cromosoma 6 (región 6P21.3, banda 21 del brazo corto del cromosoma). Los genes que forman este complejo codifican proteínas del sistema antigénico encargado del control de la respuesta inmunitaria. A nivel fisiológico, el sistema HLA está formado por moléculas de la superficie celular que regulan la respuesta inmune mediante el reconocimiento antígeno-anticuerpo por los linfocitos T. El sistema HLA del MHC está formado por dos clases de moléculas: de Clase I y de Clase II, además de por los componentes del complemento (generalmente denominados de Clase III) y otros genes productores de enzimas, como la 21-hidroxilasa, que se suelen denominar de Clase IV.
Tanto los genes de Clase III como los de Clase IV no tienen función alguna en la presentación del antígeno a los linfocitos T. El conjunto de genes HLA de Clase I y II se caracterizan por tres importantes propiedades:
- Elevado polimorfismo: permite un elevado reconocimiento peptídico.
- Codominancia alélica: se expresan las dos variantes en los heterozigotos.
- Ligamiento genético: los genes están muy próximos y casi no hay entrecruzamiento.
El elevado polimorfismo se debe a la existencia de un gran número de alelos para cada gen. Además, los dos alelos de los cromosomas homólogos se expresan al mismo tiempo (codominancia), por lo que están presentes los dos productos para cada gen en un individuo heterozigoto. Dado el pequeño tamaño del brazo corto del cromosoma 6 y la proximidad de los loci en la banda p 21.3 del cromosoma, los genes del sistema HLA tienden a transmitirse juntos (ligamiento). Sin embargo, existe la posibilidad de entrecruzamientos, especialmente entre determinadas regiones del sistema denominadas puntos calientes, pudiéndose definir combinaciones alélicas o haplotipos poblacionales en función de la combinación alélica de la que es portador un individuo.
Antígenos de Clase I
Las moléculas de Clase I son codificadas por tres loci polimórficos principales (HLA-A, HLA-C y HLA-B), aunque en la especie humana se han reconocido hasta 6 variantes génicas de Clase I: HLA-F, HLA-G, HLA-A, HLA-E, HLA-C y HLAB. Las variantes alélicas de estos genes son moléculas polimórficas que se encuentran en la membrana celular de la mayoría de las células nucleadas del organismo, aunque no se encuentran en células espermáticas, nerviosas y algunas líneas de holoblastos. Las moléculas de Clase I determinan la interacción antigénica junto con los receptores (TCR) de tipo CD8+ de los linfocitos TC citotóxicos e intervienen en los mecanismos de estimulación de la producción de anticuerpos.
Antígenos de Clase II
Los antígenos de histocompatibilidad de Clase II son moléculas que se expresan en un número limitado de células (linfocitos B, macrófagos, monocitos, linfocitos T activados pero no en reposo, células de Langerhans de la piel y células dendríticas del timo). Estas moléculas consisten en dos cadenas glicoproteicas similares, una α pesada (de 32 a 34 KD) y otra β ligera (de 29 a 32 KD), que están transitoriamente asociadas en la célula (pero no en la membrana) con una cadena invariante (li) codificada por una gen independiente situado en el cromosoma 5. Las moléculas de Clase II regulan la presentación de fragmentos peptídicos a los receptores celulares (TCR) de tipo CD4+ de los linfocitos TH cooperantes que intervienen en la activación de las células B del sistema immunológico.
Antígenos de Clase III y de Clase IV
Los antígenos de Clase III (situados entre los de clase I y II) no intervienen en el sistema de reconocimiento antígeno-anticuerpo. Se trata de los loci C2, BF, C4A (antígeno Rodgers) y C4B (antígeno Chido) que forman los denominados componentes del complemento. Estos loci pueden presentar alelos nulos, no funcionales (especialmente en los loci C4) y duplicación de alelos C4A o C4B. Además suelen presentarse asociaciones con determinados genes de Clase I y Clase II (complotipos). Los loci de antígenos de Clase III codifican componentes complementarios del suero (C2, factor B, C4A y C4B). En la región denominada de Clase IV se encuentran algunos genes del metabolismo no implicados en la función inmunitaria. La 21-hidroxilasa y un gen que controla el metabolismo del hierro son los ejemplos más importantes.
Los receptores TCR (T Cell Receptors)

Molécula de doble cadena del TCR
La principal función de las moléculas del sistema HLA es la presentación antigénica a los receptores TCR de las células T. Éstas células presentan unos 20.000 receptores a nivel de la membrana, tanto si están activadas como si no. Cada célula T expresa un solo tipo funcional de receptor TCR, formado por dos cadenas polipeptídicas (α y β o bien γ y δ codificadas por genes separados (localizados en el cromosoma 14 los loci de las cadenas α y δ, y en el cromosoma 7 los de las cadenas β y γ). Los receptores TCR forman parte de la superfamilia de genes Ig, de la que también son miembros las moléculas de Clase I y de Clase II, las moléculas CD4 y CD8, los receptores Poli-Ig y las moléculas IgM. Todas ellas participan en aspectos diversos del sistema immune. La diversificación de la especificidad de los receptores TCR se debe a la 1) diversificación de los loci codificantes de los dominios de la molécula, existiendo hasta 75 segmentos del dominio Vα, 25 del Vβ y dos del C (Cα1, Cβ1, Cα2 y Cβ2), a la 2) recombinación de los segmentos, a la 3) flexibilidad de las uniones y adiciones aleatorias, y a la 4) asociación combinatoria de las subunidades peptídicas. Esta posibilidad de variación produce hasta 108 tipos de receptores distintos, mientras que la mutación somática no ocurre en los genes TCR.
Polimorfismo del sistema HLA
El sistema HLA es altamente polimórfico. El análisis de la variabilidad alélica de los genes del sistema HLA se ha basado en gran medida en la detección serológica mediante reacciones de citotoxicidad de aloantisueros humanos en cultivos linfocitarios. Sin embargo, este sistema de detección no permite distinguir todos los tipos de alelos, ya que algunas familias alélicas producen idéntica respuesta serológica. Aun así, el número de alelos detectados serológicamente ya mostró en los años 70 y 80 el elevado polimorfismo de este sistema genético. Actualmente la tipificación alélica se realiza mediante análisis de secuencias del DNA de los genes implicados. La gran variabilidad detectada responde a múltiples factores. Por un lado, el polimorfismo del sistema MCH determina la especificidad antigénica, por lo que una elevada heterozigosidad aumentaría el número de antígenos que reconocen las moléculas del HLA y, por tanto, a los que las células T podrían responder. Por otro, el efecto de la selección natural sobre determinadas sustituciones moleculares puede detectarse comparando la frecuencia de sustituciones en regiones no codificantes con la observada en determinadas posiciones codificantes del sistema HLA. Si alguna posición ha sido seleccionada para mantener un elevado polimorfismo, la frecuencia de sustituciones no-sinónimas (determinantes de modificaciones el la secuencia de aminoácidos) aumentará respecto al de otras posiciones. El análisis molecular del DNA revela que existen regiones para los que la frecuencia de sustituciones no-sinónimas es mayor de lo esperado, coincidiendo estas regiones con las zonas de unión de los residuos peptídicos (aloantígenos) en las moléculas glicoproteicas codificadas por los genes del sistema HLA y con las zonas de interacción en los receptores TCR de los linfocitos T, por lo que se comprueba que la selección ha contribuido al mantenimiento del polimorfismo del sistema.
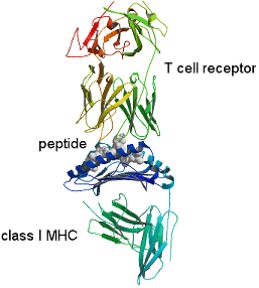
Restricción y presentación peptídica de las moléculas de Clase I y los TCR.
Las moléculas de Clase I restringen péptidos pequeños bloqueando los extremos moleculares, por lo que péptidos algo más largos simplemente “sobresaldran” en la zona central o no encajarán en la zona de restricción. Esta variación conformacional es la que determina la “modificación” de las moléculas de Clase I y facilitan su unión a los receptores TCR. En cambio, la restricción de las moléculas de Clase II mantiene libres los extremos peptídicos, lo que permite interactuar con péptidos con un mayor número de aminoácidos. Las posiciones del alopéptido que son reconocidas por las moléculas de Clase I se reducen a unos pocos aminoácidos cuyas cadenas laterales se introducen en las bolsas de las posiciones de unión. La mayoría de seres humanos son heterozigotos para los loci del sistema HLA por lo que en cada individuo se expresan seis familias de moléculas de Clase I, dos para cada alotipo HLA-A, HLA-B y HLA-C, con unas 250.000 copias de cada una presentes en la superficie celular. En función del péptido de presentación de cada alotipo las familias se subdividen en miles de subpoblaciones funcionales. Esta diversidad se corresponde con el repertorio de receptores TCR que se ha estimado en millones de formas variantes.
Diversificación filogenética
Si comparamos las variantes alélicas entre el ratón y los humanos, se comprueba que no existe correspondencia entre sus alelos individuales, por lo que la separación filogenética de 80 millones de años ha sido suficientes para reconfigurar todos los alelos de Clase I y de Clase II. En cambio al comparar los alelos humanos con los de otros primates superiores se observa que especies estrechamente emparentadas comparten una gran parte de las sustituciones nucleotídicas que forman los polimorfismos y que el grado de compartición disminuye al aumentar el grado de separación. La explicación más parsimoniosa consiste en que los alelos compartidos habrían estado presentes en el ancestro común y habrían sido heredados por ambos linajes descendientes. Algunos linajes son compartidos, por ejemplo, entre humanos y los primates del Viejo Mundo, pero no se encuentran en los del Nuevo Mundo. Sin embargo, no se han encontrado alelos idénticos entre diferentes especies de Primates, lo cual indica que los alelos son constantemente modificados a lo largo de la evolución de las especies.
Conversión alélica
La comparación alélica demuestra que existen pares de alelos que difieren tan solo en un corto segmento de su secuencia (nunca mayor de 100 nucleótidos), que es a su vez idéntico a la secuencia homóloga de un tercer alelo. Este patrón implica que la recombinación entre alelos en forma de conversión intra-alélica contribuye a la formación de nuevos alelos del mismo locus. La conversión entre alelos de diferentes loci es poco frecuente, aunque se ha detectado en genes de Clase I del ratón. El papel de la conversión en el mantenimiento del polimorfismo del sistema HLA depende del locus considerado y es muy aparente en el HLA-B. Las mutación puntual para el sistema HLAMHC explica una parte del polimorfismo, pero la recombinación por conversión se superpone al mecanismo de mutación puntual no-silenciosa. La conversión no genera nueva diversidad, pero una vez generados los alelos, la conversión puede aumentar su número generando nueva diversidad por combinación de la existente.
Selección y recombinación
Evidencias circunstanciales sugieren que la molécula antecesora de los antígenos de Clase I y II tenían una organización similar a las actuales moléculas de Clase II. Además, existe un cierto grado de conservadurismo en las moléculas de Clase II entre los mamíferos que no se observa en las de Clase I, para las que existe un alto grado de variación. El número de genes de Clase I entre los mamíferos varía entre 6 y más de 1000. El alto polimorfismo detectado se circunscribe a entre 1 y 3 genes llamados “clásicos” (de Clase Ia) que se expresan en gran número de tipos celulares, mientras que el resto son pseudogenes o genes “no clásicos” (de Clase Ib), menos polimórficos, con una distribución tisular variada. Algunos genes de Clase Ib han especializado su función de presentación del antígeno (como es el caso de la presentación de la N-formilmetionina por el gen Hmt2 de la rata), pudiendo haber derivado de la selección e loci “clásicos” de Clase I. Los genes clásicos humanos HLA-A, HLA-B y HLA-C muestran signos de especialización mediante selección, ya que presentan diferencias en cuanto a su regulación, expresión a nivel de la superficie celular o del nivel de polimorfismo. La selección balanceadora podría ser responsable del polimorfismo del sistema.
Dos son los mecanismos de selección que se han sugerido para explicar el elevado polimorfismo del sistema: 1) ventaja selectiva del heterozigoto, y 2) selección dependiente de las frecuencias. La selección positiva del heterozigoto vendría dada por la ventaja adaptativa (de supervivencia y reproducción) de presentar inmunidad ante un elevado número de agentes patógenos o parasitarios. La heterozigosis permitiría la presentación antigénica de hasta el doble de péptidos que un individuo homozigoto, resultando en una respuesta inmunitaria más fuerte. El éxito de la heterozigosis depende del número de alotipos que se expresen, por lo que la codominancia alélica marcará la diferencia en cuanto al número de familias moleculares expresadas. Este efecto es evidente en las seis familias de alelos del HLA-A, para el que casi todos los humanos somos heterozigotos. Esta selección del heterozigoto es indiferente e independiente de las demandas del parásito. Una población que se enfrente a una infección epidémica y una reducción poblacional severas, estará sometida a una fuerte presión selectiva bajo la acción de un sólo agente parasitario. Cualquier alelo que por azar provea de una mayor inmunidad frente a ese agente verá aumentada su frecuencia rápidamente. Por ejemplo, La creciente frecuencia del alelo HLA-B*3501 en la población de Gambia parece ser el resultado directo de la selección causada por el parásito de la malaria y la enfermedad que causa. La frecuencia de un alelo seleccionado positivamente podría aumentar en la población hasta hacerse mayoritario. Sin embargo, los parásitos también pueden adaptarse a los alotipos del huésped por mutación de las secuencias peptídicas a las que el sistema inmunitario responde. Este tipo de adaptación iría dirigido contra los alelos más frecuentes en la población huésped, proporcionando una ventaja selectiva a los más raros. Así, a medida que un alelo seleccionado positivamente aumenta su frecuencia en una población, se convierte en la diana más probable para la adaptación del parásito.
La selección mediante parásitos no es la única fuerza evolutiva de los genes de Clase I. La recombinación es el segundo factor en importancia. La evolución de los genes de copia única se produce por la acumulación progresiva de mutaciones puntuales a partir del tipo salvaje, lo cual permite establecer filogenias, como es el caso de alelos del mt-DNA donde no existe recombinación. Sin embargo, los genes de Clase I no responden a estas reglas. Una gran variedad de mecanismos de recombinación actúan alterando la diversidad generada por las mutaciones puntuales. El entrecruzamiento desigual puede duplicar o eliminar genes; la recombinación y la conversión alélica redistribuyen y mezclan las sustituciones que aparecen en alelos y genes individuales. Por tanto, la interacción entre los factores de mutación, selección diversificadora, conversión génica y deriva genética es importante para la adquisición y mantenimiento del polimorfismo en los genes de Clase I.
Desequilibrio por ligamiento: haplotipos
Una característica importante del sistema HLA es la proximidad entre los distintos loci que produce un ligamiento entre los caracteres (linkage desequilibrium). Algunos alelos aparecen juntos con mayor probabilidad que la que cabría esperar, manteniéndose estables sus frecuencias de generación en generación. Este equilibrio se puede romper por el 1) efecto fundador, por 2) fusión poblacional o por 3) selección natural. Sin embargo, el desequilibrio tiende a desaparecer en ausencia de selección en poblaciones con cruzamientos aleatorios. Se ha calculado que con un coeficiente de recombinación del 0.8% entre los loci HLA-A y HLA-B, en 200 generaciones (unos 5.000 años) se reduciría el desequilibrio en un factor de 5. Los loci HLA-B y HLA-C, más próximos entre si con un factor de recombinación de 0.2%, necesitarían unos 20.000 años de entrecruzamientos aleatorios para reducir en 5 veces el desequilibrio por ligamiento.

- Oceanía: Dado que las primeras poblaciones austronesias llegaron a Oceanía hace no más de 5.000 años tendrían que encontrarse aún evidencias de desequilibrio por ligamiento producidos por el contacto entre las poblaciones Papúes (australomelanesias) y austronesias. La combinación alélica más discriminante en poblaciones de Oceanía es el haplotipo HLA-B13-Cw4. Las frecuencias observadas en poblaciones de Australia, Nueva Guinea (costa e interior) y Nueva Caledonia son significativamente distintas a las esperadas, lo que sería indicativo de un origen común, distinto del de poblaciones polinesias, en las que el alelo HLA-B13 se ha perdido y el Cw4 se mantiene con una baja frecuencia. Así, el HLA parece apoyar la visión de que:
- Las poblaciones de Australia y de las tierras altas de Nueva Guinea tendrían un ancestro común de gran antigüedad.
- Las poblaciones costeras de Nueva Guinea y de las islas de Melanesia comparten caracteres y afinidades con un componente aproximado del 15% de herencia austronésica (en torno al 20% en Fiji).
- Los polinesios habrían tenido poco contacto con micronesios, a pesar de la similitud en la apariencia física, con un origen separado de los Micronesios (sugerido por el HLA).
- Aunque los elementos austronésicos influenciaron a las poblaciones melanesias, no hubo influencia melanesia en Polinesia, que probablemente fué poblada por un bajo efectivo poblacional, como sugiere la ausencia de algunos alelos com HLA-B13 y B27.
- El sistema HLA sugiere que las afinidades entre el Este de Polinesia y las poblaciones americanas son antiguas, anteriores a la divergencia de este grupo en el Sud-Este asiático.
- América: La distribución de frecuencias de alelos de Clase I del sistema HLA apoyan el escenario mas generalmente aceptado en el que los pobladores de America tendrían un origen asiático, cruzando por el estrecho de Bering entre hace 10.000 y 40.000 años. En las poblaciones amerindias actuales encontramos alelos únicos de polimorfismos de la Clase I. Estas poblaciones tienen mucha menos cantidad de alelos HLA-A, -B y -C, derivados todos ellos de 4 alelos HLA-A, 9 -B y 7 -C, que comparten con las poblaciones asiáticas y europeas. Estos alelos compartidos serian probablemente los que estaban presentes en las poblaciones fundadoras del nuevo mundo. De todas formas, hay diferencias notables entre el panorama en Norteamérica y Sudamérica, existiendo en el sur mucha más variabilidad y mayor frecuencia de “nuevos” alelos. Así, se observa que:
- Durante un periodo comparable de tiempo, el total de alelos de HLA de Clase I permaneció constante en el norte y varió en el sur.
- Los tres loci -A,-B y -C han evolucionado de diferente forma y a diferente velocidad en America.
- La recombinación produce nuevos alelos de forma mucho más rápida que la substitución puntual. Estas conclusiones no son exclusivas de las poblaciones
amerindias.
 Riesgo relativo
Riesgo relativo
Cabe esperar que las moléculas del HLA estén asociadas a la protección inmunológica frente a determinadas enfermedades infecciosas. Sin embargo, en ocasiones los alelos del HLA se asocian con elevada probabilidad de padecer ciertas enfermedades. Es lo que se conoce como riesgo relativo, que se define como el cociente entre la probabilidad de padecer una determinada enfermedad en ausencia del alelo del HLA y la probabilidad de padecerla en presencia de ese mismo alelo:
- Riesgo relativo = P[enfermedad/sin-alelo-HLA] / P[enfermedad/con-alelo-HLA]
Valores inferiores a 1 indican que el alelo del HLA confiere resistencia a dicha enfermedad, mientras que valores mayores a 1 indican que tener dicho alelo confire un riesgo de padecer la enfermedad.
 Algunas enfermedades muestran un elevado riesgo relativo, come el riego de padecer narcolepsia en presencia del alelo DR2, que es de 130 (se eleva 130 veces el riesgo de padecer la enfermedad en presencia de ese alelo). En cambio, el alelo B53 parece conferir resistencia a la malaria, con un riesgo relativo de 0,59. Esta asociación entre determinados alelos del HLA y algunas enfermedades se explica por una posible asociación de proximidad (ligamiento) en el cromosoma con algún determinante genético directamente implicado con la enfermedad. Cabe esperar, por tanto, que la selección actúa a dos niveles: 1) favoreciendo alelos del HLA por conferir ventaja en el reconocimiento inmunológico de alguna enfermedad infecciosa y 2) contraselección de alelos del HLA por asociación con algunas enfermedades genéticas aociadas por ligamiento genético.
Algunas enfermedades muestran un elevado riesgo relativo, come el riego de padecer narcolepsia en presencia del alelo DR2, que es de 130 (se eleva 130 veces el riesgo de padecer la enfermedad en presencia de ese alelo). En cambio, el alelo B53 parece conferir resistencia a la malaria, con un riesgo relativo de 0,59. Esta asociación entre determinados alelos del HLA y algunas enfermedades se explica por una posible asociación de proximidad (ligamiento) en el cromosoma con algún determinante genético directamente implicado con la enfermedad. Cabe esperar, por tanto, que la selección actúa a dos niveles: 1) favoreciendo alelos del HLA por conferir ventaja en el reconocimiento inmunológico de alguna enfermedad infecciosa y 2) contraselección de alelos del HLA por asociación con algunas enfermedades genéticas aociadas por ligamiento genético.
Técnicas de análisis de la diversidad molecular
Los Polimorfismos de DNA que afectan a porciones del genoma codificador de proteínas constituye tan solo el 5%-10% del polimorfismo genómico total. Dos copias cualesquiera del genoma humano difieren por término medio en 1 posición de entre 100 ó 500. Así, si se pudieran comparar directamente todas posiciones del genoma haploide de dos progenitores, diferirían en más de 10 millones de posiciones, producidas por acumulación de mutaciones a lo largo de la evolución del ser humano. A partir de sondas de DNA se ha calculado que existiría 1 base polimórfica por cada 270 posiciones nucleotídicas tomadas al azar (índice de heterozigosis). Esta cifra es 10 veces superior a la proporción de nucleótidos heterozigotos de las regiones codificadoras (1 de cada 25.000 bases). Ello se debe a que las regiones codificantes se hallan bajo una presión selectiva muy intensa y, por tanto, la incidencia de mutaciones en estas zonas es menor de la que cabría esperar. Los polimorfismos de DNA son la manifestación molecular de la variación en el genoma. La detección de los polimorfismos de DNA se realiza mediante diversas técnicas de análisis (Thompson & Thompson, 1996).
Polimorfismos de longitud de restricción de fragmentos (RFLP)
Los RFLPs (Restriction Fragment Length Polymorphism) se deben a variaciones en las secuencias nucleotídicas del DNA que no comportan variaciones en el fenotipo. Los RFLPs pueden ser debidos a: 1) mutaciones puntuales, 2) pequeñas delecciones o inserciones de secuencias también pequeñas, o a 3) repeticiones de secuencias. Estas variaciones son frecuentes y ocurren de forma aleatoria a lo largo de todo el genoma humano (1 por cada 200 pares de bases por término medio). Se pueden poner de manifiesto mediante enzimas de restricción que cortan el DNA en dianas específicas. La presencia o ausencia de una o varias dianas por efecto del polimorfismo produce fragmentos de DNA de tamaño diverso en cada persona en función de su secuencia genómica particular. Los fragmentos de restricción afectados por los polimorfismos presentan una movilidad electroforética diferente según su tamaño. Cada RFLP se hereda mediante codominancia mendeliana simple. Por ello, es posible interpretar directamente las longitudes de los fragmentos de RFLP como un reflejo del genotipo (secuencia del DNA) en un lugar de restricción determinado. Una delección o una inserción también modifican el tamaño del fragmento de DNA considerado sin necesidad de utilizar enzimas de restricción.
- El gen de la α1-antitripsina: La α1-AT es una proteína sérica que inhibe la actividad de varios enzimas proteolíticos, como la tripsina, la quimotripsina y la elastasa pancreática. Su diana principal es la elastasa leucocitaria que en ausencia de α1-AT destruye las proteínas del tejido conjuntivo pulmonar, en particular la elastina, dañando la pared alveolar. El gen de la α1-AT es muy polimórfico y se conoce como gen PI (Inhibidor de la proteasa) y se encuentra en el cromosoma 14. Existen tres alelos frecuentes (M1, M2, M3) que codifican proteínas normales, aunque son estructuralmente diferentes. Otros alelos codifican proteínas con actividad muy reducida del inhibidor de la proteasa. El alelo Z (G→A, Glu→Lys) tiene una frecuencia del 1% al 2% en poblaciones de raza blanca y en homozigosis ZZ produce una concentración de α1-AT en plasma menor al 15% de la concentración normal, con riesgo elevado de enfermedad pulmonar (en la edad adulta temprana). El alelo S (A→T, Glu→Val) también codifica productos con actividad reducida, pero es menos severo (concentración del 60% de lo normal). La deficiencia de α1-AT se encuentra con una frecuencia de 1/2000 a 1/8000 en poblaciones de raza blanca y es rara en poblaciones de raza negra o asiática. El gen de la α1-AT presenta un alto grado de variabilidad molecular de DNA, incluyendo las mutaciones que producen alelos patológicos (Z, S). Algunas variantes se han detectado por RFLPs y sus frecuencias varían de una población a otra.
- DNA satélite: Tan solo ¾ partes del genoma esta formado por DNA de copia única y el resto lo forma el DNA repetitivo, cuya función podría estar relacionada con el mantenimiento de la estructura cromosómica. Sin embargo, se desconoce exactamente la utilidad del DNA repetitivo. Se han identificado varias categorías de este DNA repetitivo que se diferencian en si las repeticiones se agrupan en una o varias posiciones o se entremezclan con fragmentos de DNA de copia simple (asociados a loci codificantes). Entre las secuencias repetitivas cabe destacar las denominadas secuencias Alu que son repeticiones de DNA que pueden llegar a ser de gran tamaño. A diferencia de las secuencias Alu, los minisatélites están formados por repeticiones de nucleótidos de sólo 15 a 65 pares de bases, generalmente formando fragmentos de hasta unas 20 kb aproximadamente que se distribuyen a
lo largo de todo el cromosoma. La detección de este tipo de polimorfismo se realiza mediante detección de longitud de fragmentos sin necesidad de restricción enzimática.  Anemia Falciforme: la anemia falciforme se debe a una mutación en la cadena β de la Hemoglobina normal (alelo A) produciendo una sustitución (alelo S) de una base (Adenina por Timina). Este cambio en un aminoácido produce una alteración en la estructura terciaria de la molécula que causa una alteración en la morfología de los glóbulos rojos. Esta mutación se puede detectar mediante una diana de restricción enzimática que permite amplificar fragmentos de diferente tamaño en homozigotos y heterozigotos, facilitando la caracterización del genotipo en individuos portadores de la mutación respecto a los no portadores.
Anemia Falciforme: la anemia falciforme se debe a una mutación en la cadena β de la Hemoglobina normal (alelo A) produciendo una sustitución (alelo S) de una base (Adenina por Timina). Este cambio en un aminoácido produce una alteración en la estructura terciaria de la molécula que causa una alteración en la morfología de los glóbulos rojos. Esta mutación se puede detectar mediante una diana de restricción enzimática que permite amplificar fragmentos de diferente tamaño en homozigotos y heterozigotos, facilitando la caracterización del genotipo en individuos portadores de la mutación respecto a los no portadores.
Número Variable de Repeticiones en Tandem (VNTR)

Fingerprinting.
Una clase particular de polimorfismo de inserción o delección de fragmentos de DNA es el llamado polimorfismo VNTR (Variable Number of Tandem Repeats). Son repeticiones variables de fragmentos de DNA en tandem, generalmente repeticiones de dos o tres pares de bases. Se trata de un polimorfismo hipervariable ya que los loci de las repeticiones se caracterizan por presentar múltiples alelos, hasta varias decenas o más. Los VNTR son, por tanto, buenos indicadores de paternidad, ya que es poco probable que dos individuos no emparentados compartan exactamente los mismos alelos, y son también de utilidad en el estudio del ligamiento genético. Su detección se realiza mediante hibridación con una sonda marcada, externa al la zona de las repeticiones, y gel de resolución por diferencias en tamaño entre los distintos alelos. En la actualidad se conocen varios miles de polimorfismos, tanto de RFLP como VNTR (ambos tipos presentan herencia codominante). Los STR (Short Tandem Repeats) son repeticiones en tandem de pequeños fragmentos de DNA, de 3 a 15 pares de bases generalmente), que se detectan de la misma forma que los VNTRs.
- Fingerprinting (huella digital de DNA): Muchos polimorfismos de VNTR que
comparten secuencias repetidas o son suficientemente similares que pueden ser detectados simultáneamente, utilizando como sonda una secuencia repetida común. Esto resulta en un patrón de bandas que es específico y único de cada individuo, por lo que constituye su “huella digital” de DNA. Sólo los gemelos idénticos presentan patrones indistinguibles.
Polimorfismos nucleotídicos únicos (SNPs)
Los SNPs (Single Nucleotide Polymorphisms) se detectan por secuenciación de DNA. Permite detectar el máximo nivel de variabilidad. Se aplica en particular a zonas
no codificantes, como la Región de Control del DNA mitocondrial.

DNA mitocondrial

mtDNA (© Emmanuel Douzery)
El mtDNA es un DNA circular con 16,569 pares de bases que codifica 37 genes (2 ARN ribosómicos, 22 ARN de transferencia y 13 proteínas que participan en la fosforilación oxidativa). Se hereda por vía materna; cuando un espermatozoide fecunda un óvulo penetra el núcleo y su cola junto con sus mitocondrias son destruidos en el óvulo materno. Por lo tanto, en el desarrollo del cigoto sólo intervendrían las mitocondrias contenidas en el óvulo. Sólo en algunos casos las mitocondrias del espermatozoide pueden ingresar al óvulo. Además el mtDNA no presenta entrecruzamiento. El ADN mitocondrial codifica 13 proteínas involucradas en la producción de energía celular y procesos de fosforilación oxidativa. Por lo tanto, el entorno que rodea la mitocondria y el ADN mitocondrial está expuesto al daño oxidativo producido por los radicales libres generados en ese metabolismo. Además, el material genético de las mitocondrias no está protegido por histonas como lo está el ADN nuclear, y los mecanismos de reparación de daños del ADN son poco eficientes en las mitocondrias, por lo que la tasa de mutación es hasta 10 veces mayor que en el genoma nuclear Por ello, el mtDNA es altamente polimórfico en aquellas regiones no sujetas a selección natural (zonas codificantes de genes conservativos) y se utiliza para establecer filogenias en las poblaciones humanas.
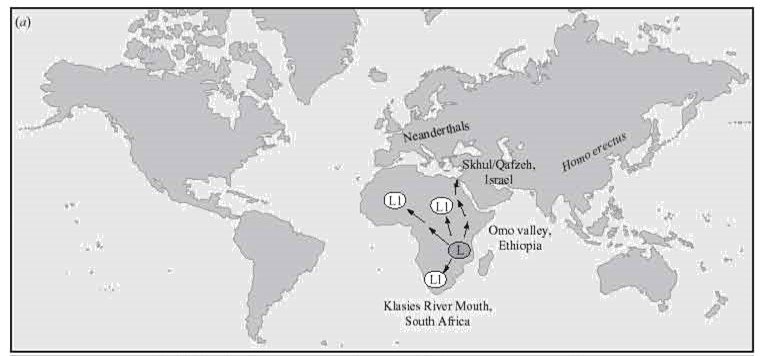
 La diversidad del ADN mitocondrial permite caracterizar la ascendencia matrilineal a partir de la acumulación de mutaciones (generalmente puntuales o SNPs) en las regiones que no codifican proteínas. A partir de la diversidad actual es posible determinar la secuencia del ancestro común de los linaje modernos, al que se ha denominado Eva mitocondrial. A la Eva mitocondrial se le ha dado una antigüedad promedio de 190.000 años y el lugar en que vivió podría coincidir con el de la mayor diversidad genética mitocondrial, que se encuentra en el en Tanzania, en África oriental. Ese linaje ancestral se ha denominado linaje L, del que deriva el linaje Lo, el más antiguo detectado en África, y los linaje L1, L2 y L3.
La diversidad del ADN mitocondrial permite caracterizar la ascendencia matrilineal a partir de la acumulación de mutaciones (generalmente puntuales o SNPs) en las regiones que no codifican proteínas. A partir de la diversidad actual es posible determinar la secuencia del ancestro común de los linaje modernos, al que se ha denominado Eva mitocondrial. A la Eva mitocondrial se le ha dado una antigüedad promedio de 190.000 años y el lugar en que vivió podría coincidir con el de la mayor diversidad genética mitocondrial, que se encuentra en el en Tanzania, en África oriental. Ese linaje ancestral se ha denominado linaje L, del que deriva el linaje Lo, el más antiguo detectado en África, y los linaje L1, L2 y L3.
 Los primeros estudios de la diversidad de linajes mitocondriales realizados por Rebecca Cann mostraron que los linaje africanos presentaban una diversidad mucho mayor que en otras zonas geográficas, incluyendo linajes exclusivos de África y otros presentes también en otras zonas. Esta mayor diversidad se interpreta como una mayor tiempo de aislamiento desde la divergencia del ancestro común (Eva mitocondrial), Dado que en el mtDNA no hay entrecruzamiento, las mutaciones presentes en su secuencia se hereda juntas y forman, por tanto, un haplotipo. El haplotipo se define como una combinación de mutaciones puntuales que se heredan juntas. Los individuos portadores de un mismo haplotipo pertenecen al mismo haplogrupo. Por consenso, lo haplogrupos ancestrales y reciente del continente africano se identifican con la letra L, utilizándose subíndice para designar los linajes derivados (Lo, L1, L2 y L3). Del L3 derivan los primeros encontrados fuera de África: los haplogrupos M y N. Los haplotipos M y N son los más antiguos fuera de África y se diversifican a medida que las poblaciones humanas se expanden por Asia, Europa y América.
Los primeros estudios de la diversidad de linajes mitocondriales realizados por Rebecca Cann mostraron que los linaje africanos presentaban una diversidad mucho mayor que en otras zonas geográficas, incluyendo linajes exclusivos de África y otros presentes también en otras zonas. Esta mayor diversidad se interpreta como una mayor tiempo de aislamiento desde la divergencia del ancestro común (Eva mitocondrial), Dado que en el mtDNA no hay entrecruzamiento, las mutaciones presentes en su secuencia se hereda juntas y forman, por tanto, un haplotipo. El haplotipo se define como una combinación de mutaciones puntuales que se heredan juntas. Los individuos portadores de un mismo haplotipo pertenecen al mismo haplogrupo. Por consenso, lo haplogrupos ancestrales y reciente del continente africano se identifican con la letra L, utilizándose subíndice para designar los linajes derivados (Lo, L1, L2 y L3). Del L3 derivan los primeros encontrados fuera de África: los haplogrupos M y N. Los haplotipos M y N son los más antiguos fuera de África y se diversifican a medida que las poblaciones humanas se expanden por Asia, Europa y América.

Diversificación de los linajes del mtDNA.
El linaje Lo se habría originado hace unos 194.000 años y la diversificación de L1 en África se habría producido hace unos 142.000 años.
Los haplotipos L2 y L3 habrían aparecido hace en torno a 132.000 años y L3 tendría una antigüedad de 96.000 años, del que derivan los haplotipos M y N son los más antiguos fuera de África.
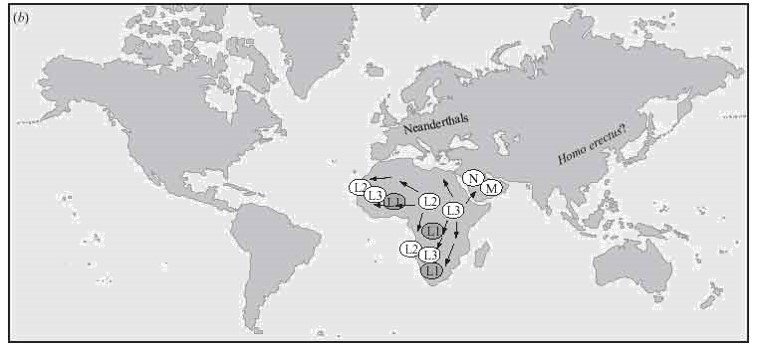
A partir del los linajes N y N se formaron linajes derivados que se expandieron por toda la geografía a la vez que se desplazaban las poblaciones humanas. Los linajes M y N, y sus derivados, se encuentran en el Sudeste Asiático y en Australia. Los linajes derivados A, B y D se encuentran en Asia, mientras que R (derivado del N) dio lugar a los haplogrupos mayoritarios en Europa (H, I, J, U, V).
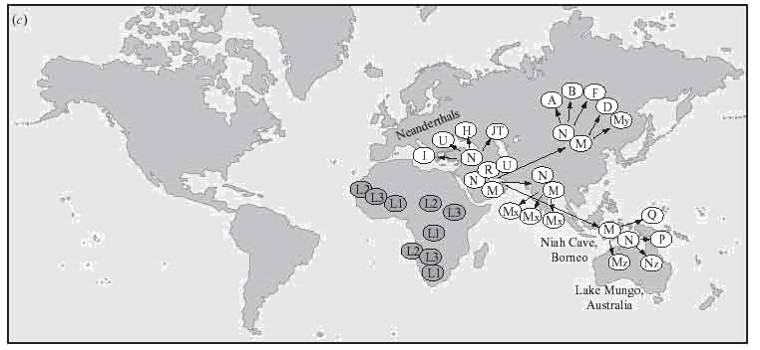
Finalmente, los linajes asiáticos A, B, C y D entraron en el continente americano, donde también se encuentra en poblaciones amerindias el linaje X, de origen europeo (probablemente introducido por incursiones de viquingos antes del descubrimiento de américa por Colón en 1492.

Bibliografía
- Cann RL (1992) The search for Eve. Science Apr 3; 256(5053), 79.
